2. 研究进展
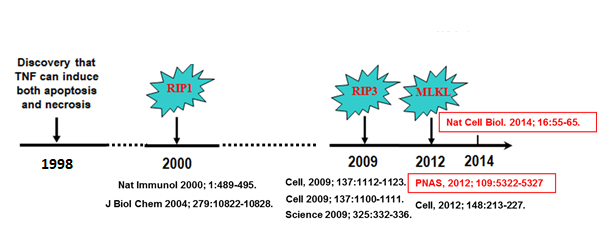
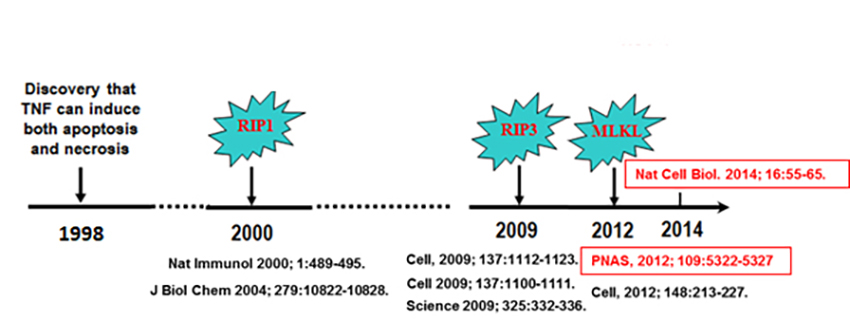
图1,坏死性凋亡信号途径的发现过程。1998年首次报道TNF-α刺激能诱发某些细胞发生坏死性死亡。2000年,坏死性凋亡的第一个关键因子-RIPK1被发现。2009年,坏死性凋亡的第二个关键因子-RIPK3被三个不同的研究小组所发现。2012年,坏死性凋亡的第三个关键因子-MLKL被发现。2014年, MLKL介导坏死性凋亡的关键机制被揭示。其中,我们课题组前期所发表的文章用红色标注。
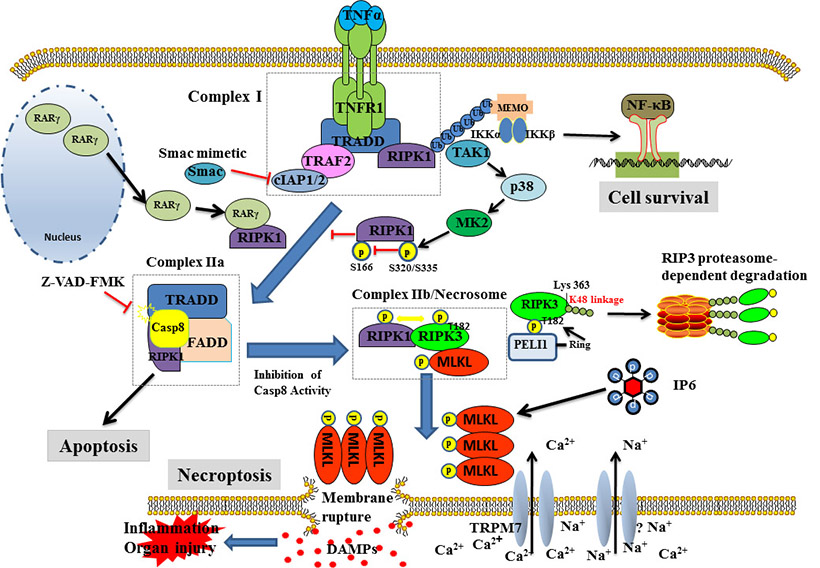
图2,坏死性细胞凋亡信号转导途径。TNF-α被其受体TNF-R1识别导致形成称为复合物I的膜信号复合物,该复合物包括TRADD、RIPK1、TRAF2和cIAP1/cIAP2。复合物激活NF-κB信号通路,从而促进细胞存活和炎症。cIAP1/cIAP2对RIPK1的多泛素化是激活NF-κB信号通路的关键。当cIAP1/cIAP2被SMAC或SMAC模拟化合物抑制时,RIPK1被去泛素化。然后,复合物I被释放到细胞质中,招募FADD形成复合物II。FADD是诱导Caspase-8二聚体化和激活的必要条件,Caspase-8执行凋亡过程。在这种情况下,RARγ与RIPK1相互作用,促进其与复合物I的解离。MK2是RIPK1依赖性细胞死亡的抑制剂。MK2被p38/MAPK信号途径激活,并在S320/S335残基处直接磷酸化RIPK1。这种磷酸化抑制了在残基S166处的RIPK1的自磷酸化,从而阻止了复合物II的形成。z-VAD-FMK对Caspase-8活性的抑制使RIPK3重新募集到复合物II形成Necrosome。Necrosome的形成将死亡信号从凋亡转变为坏死。Peli1是磷酸化RIPK3的K48泛素连接酶。其通过蛋白酶体途径诱导RIPK3降解,抑制坏死。然后,MLKL被招募到Necrosome,并被RIPK3磷酸化,导致其寡聚和质膜定位。MLKL的质膜易位是导致质膜破裂的重要因素,其可通过直接形成孔结构或激活阳离子通道来增加Ca2+和Na+离子的流入,从而提高细胞渗透压,最终使质膜破裂。当质膜破裂时,细胞质中的炎症物质释放,导致组织炎症和器官损伤。引自课题组发表综述:Biochim Biophys Acta Rev
Cancer, 1871:259-266.
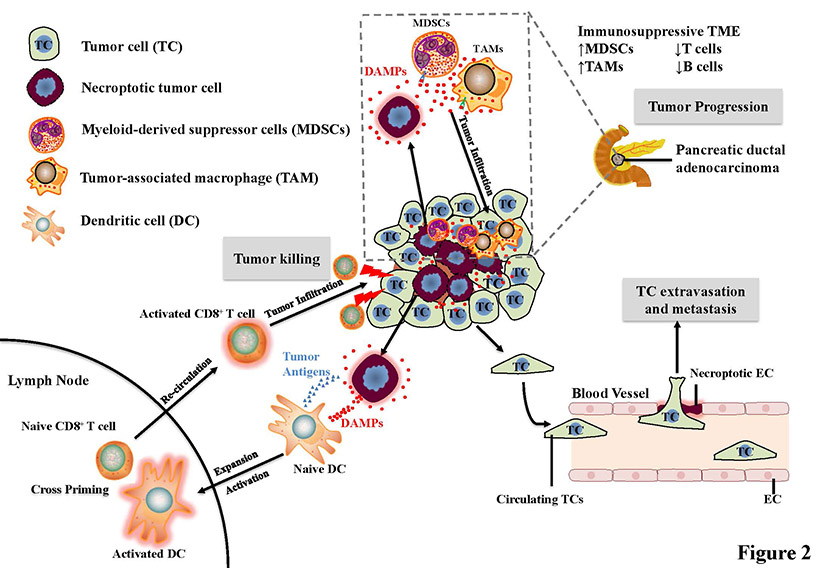
图3, 坏死性细胞凋亡的促肿瘤和抗肿瘤作用。在胰腺导管癌(PDA)中,发生坏死性凋亡的肿瘤细胞释放的DAMPs或细胞因子召集髓系来源的抑制细胞(MDSCs)和肿瘤相关巨噬细胞(TAM)诱导免疫抑制性的肿瘤微环境。这种免疫抑制性的肿瘤微环境促进肿瘤生长和进展。此外,循环血中的肿瘤细胞(TCs)诱导内皮细胞(ECs)发生坏死性凋亡,使得肿瘤细胞从坏死的内皮细胞层渗出,从而促进肿瘤转移。另一方面,发生坏死性凋亡的肿瘤细胞释放的DAMPs也能吸引树突状细胞(DCs)到达肿瘤部位,活化的树突状细胞摄取肿瘤抗原与淋巴结内的原始CD8+ T细胞交叉递呈。然后,原始CD8+ T细胞分化为细胞毒性T细胞(CTLs),浸润肿瘤部位,杀伤肿瘤细胞。引自课题组发表综述:Biochim Biophys Acta Rev Cancer, 1871:259-266.
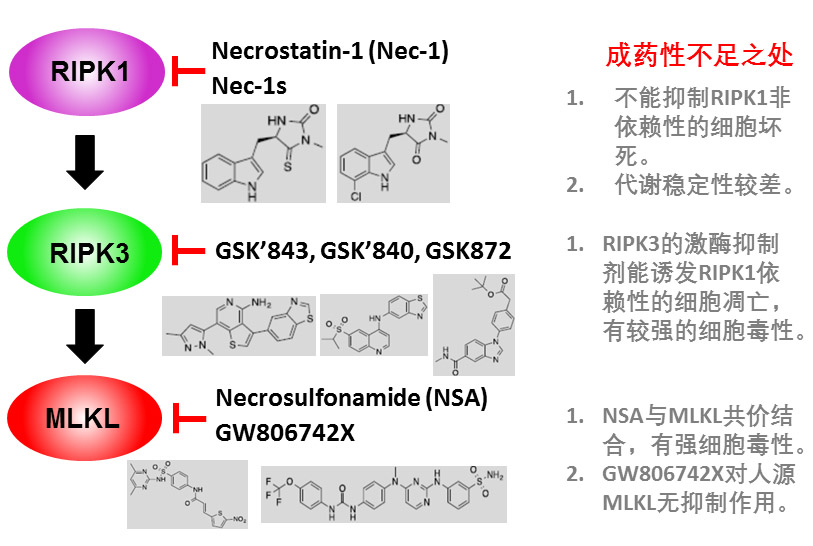
图4,当前靶向坏死性细胞凋亡信号途径关键蛋白的经典小分子的抑制剂以及它们的成药性不足之处。目前发现的小分子抑制剂存在靶点选择性弱、毒性大、体内活性弱等缺点。因此,到目前为止很少有抗坏死性凋亡小分子药物进入临床实验研究。例如,坏死性凋亡第一个被报道的小分子抑制剂Necrostatin-1 (Nec-1)是RIPK1激酶抑制剂。目前已有大量证据表明Nec-1可以通过抑制多种疾病模型中的坏死性凋亡而降低这些疾病的致死率。但是,由于其代谢稳定性较差,到目前为止Nec-1也只是作为一种工具化合物在使用。又如,RIPK3 的抑制剂GSK’840、GSK’843、GSK’872 在抑制坏死性凋亡的同时却能诱发RIPK3介导的细胞凋亡,因此具有较大的细胞毒性。